Steelhead trout, Oncorhynchus mykiss, are fish that live in streams in North America.
To increase the number of steelhead trout, captive breeding has occurred since 1992. Fish eggs and sperm are mixed and the young fish grow in large tanks of aerated water for the first year of their lives. Most are then released into the wild, however a few male and female fish are kept to become the parents of the next generation of captive-bred fish.
Each tank may hold up to 50000 fish. The young captive fish are fed processed food. Some young fish are unable to survive these conditions and a proportion die. Death is usually the result of poor wound-healing after accidents due to overcrowding and due to the spread of diseases.
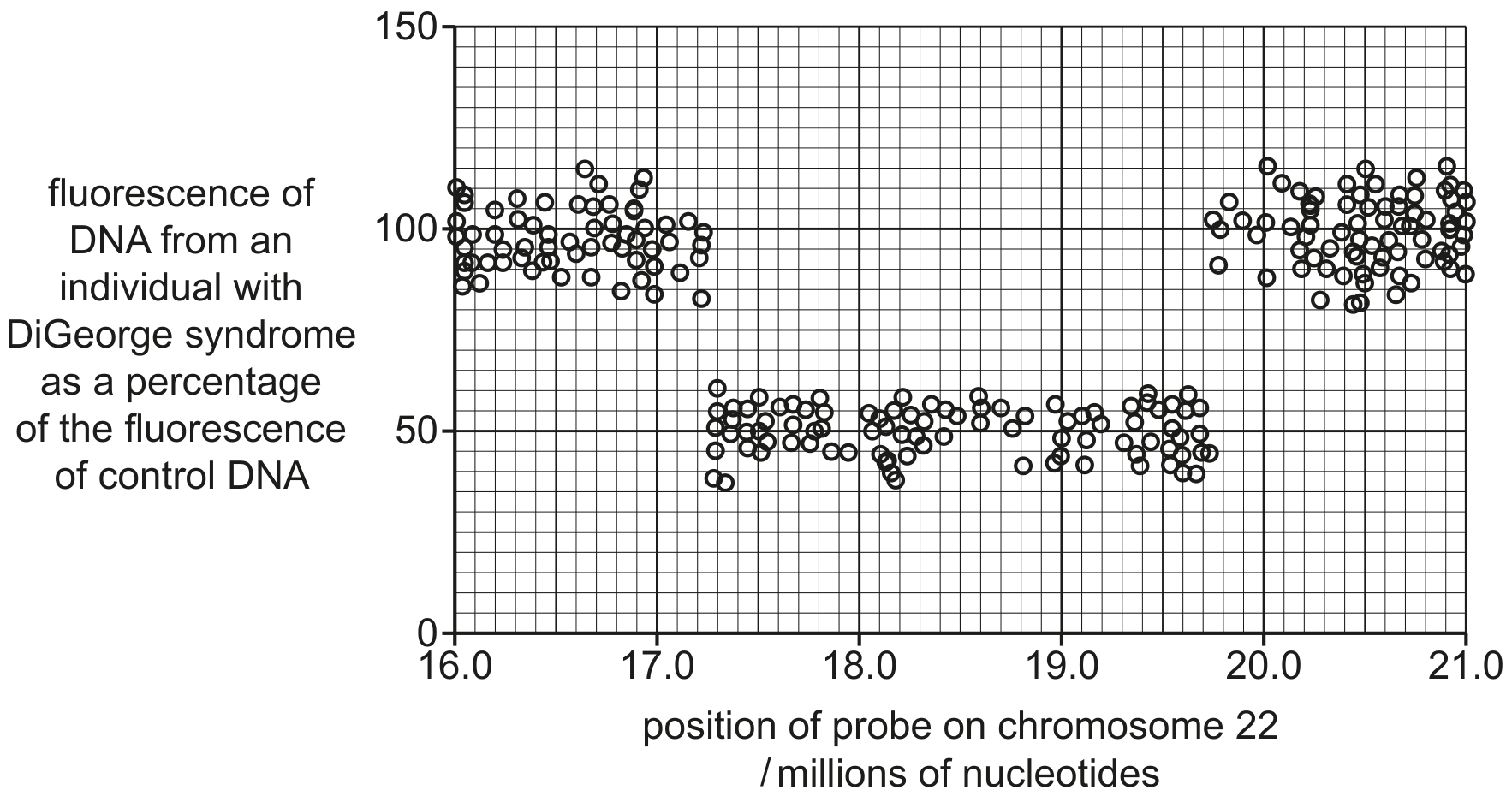
Describe how microarray analysis can detect differences in the expression of many genes when comparing two samples, such as the offspring of wild and captive-bred fish.